
tutorial
Overview of CavityPlus
CavityPlus is a web server for precise and robust protein cavity detection and functional analysis. With protein 3D structural information as input,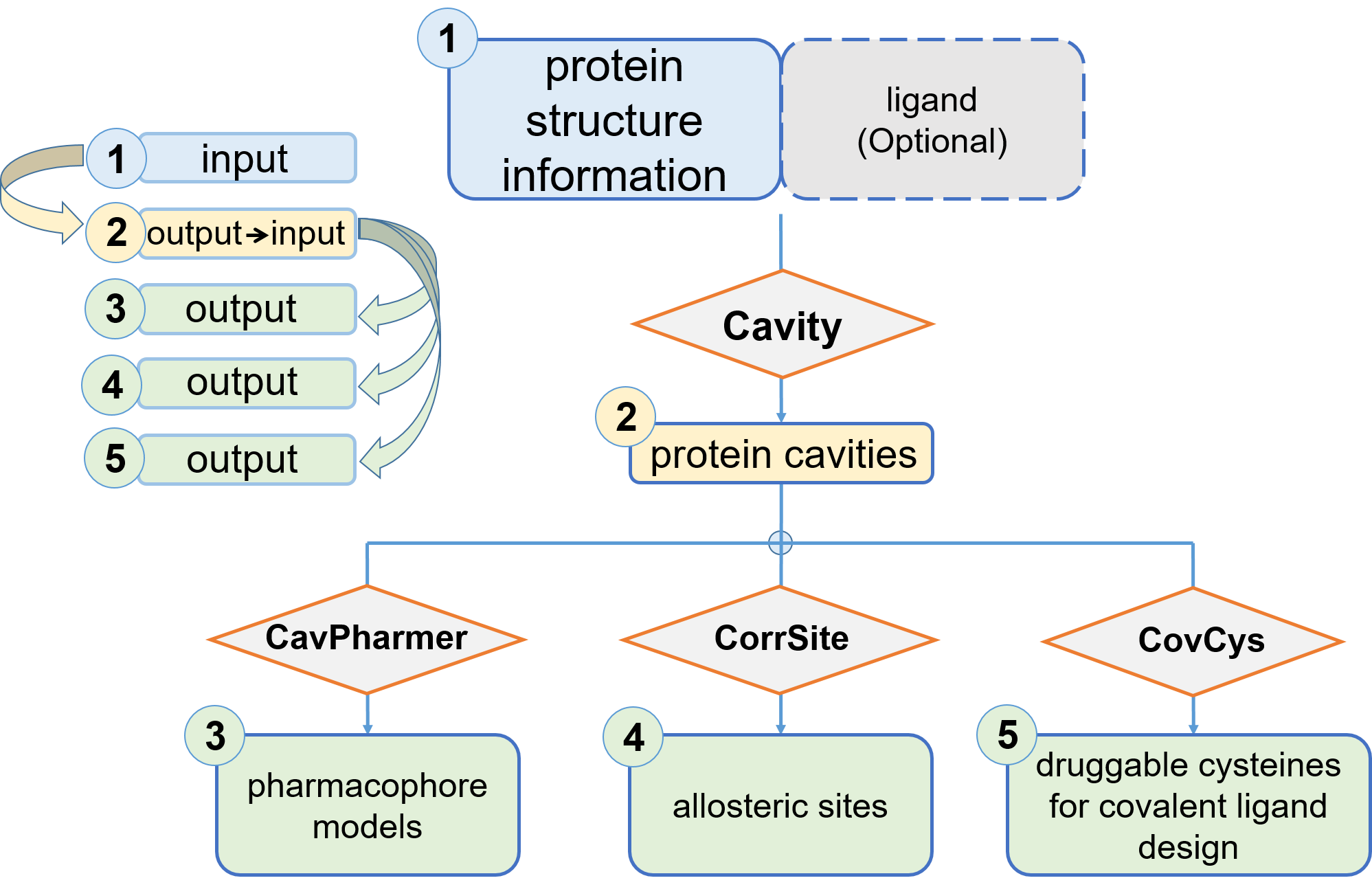
Cavity
Step 1
Load a protein of your interest. Two ways are provided here:1) Load from RCSB PDB based on a valid PDB ID;
2) Upload your own protein of interest.
After this step, one protein structure will be shown in the JSmol window.
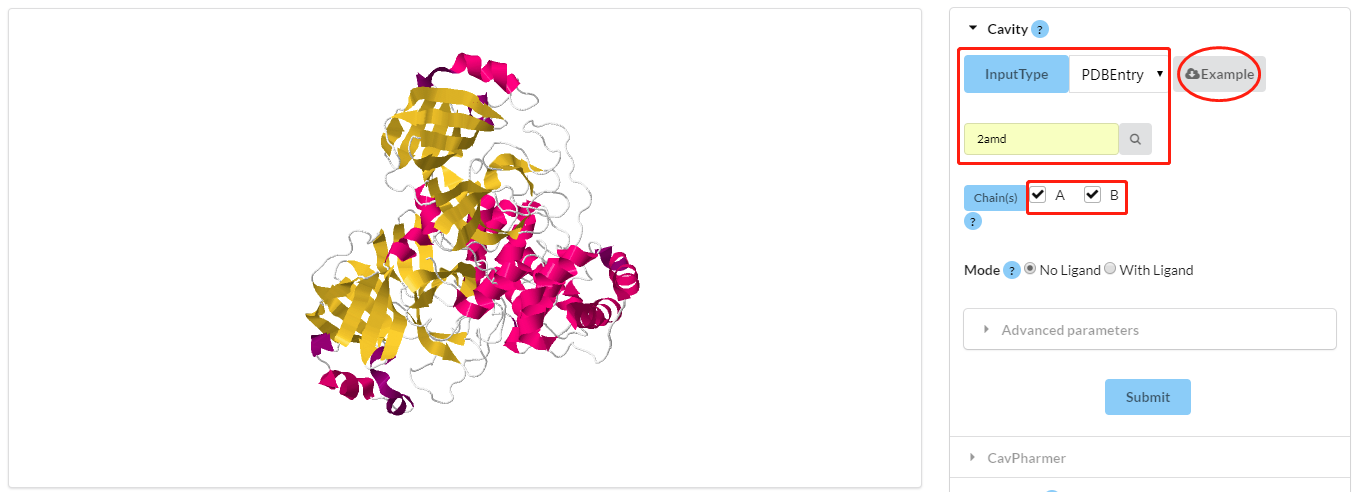
Figure 1. Visualization of Cavity operation interface. Red rectangles show the input protein and its chains. Red ellipse with the "sample" can be clicked to download.
Step 2
Select the chain(s) from the whole protein, and it will be shown in JSmol right now.
Note: CAVITY will automatically remove water molecules, ion atoms and ligand atoms from
structure.
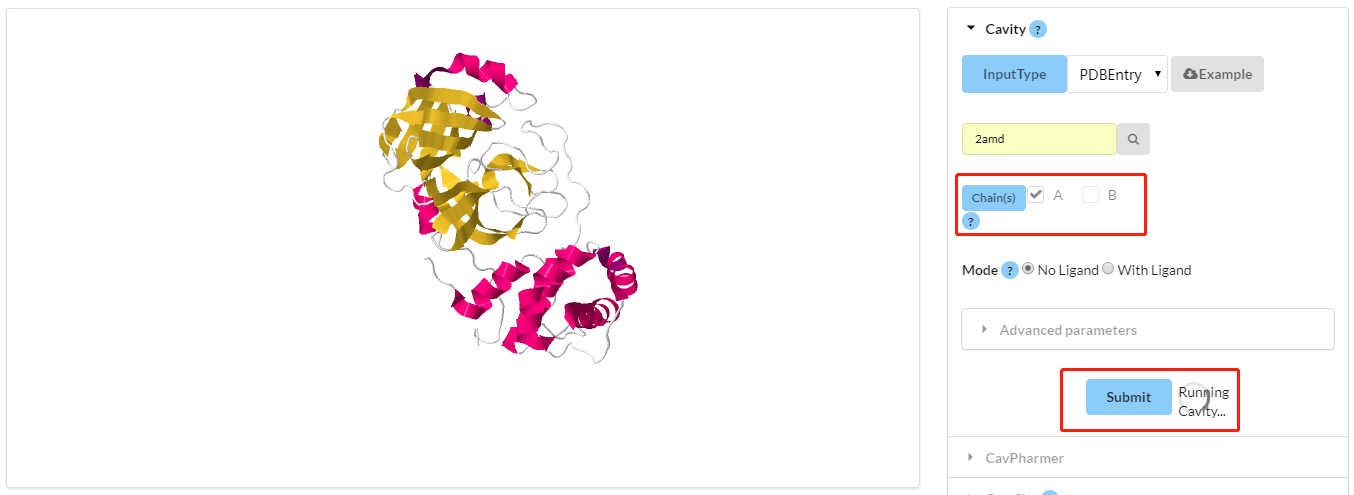
Figure 2. Visualization of Cavity submission. Red rectangles show the current selected
chain, then submit Cavity to detect pockets with a roughly estimated computational time.
Step 3
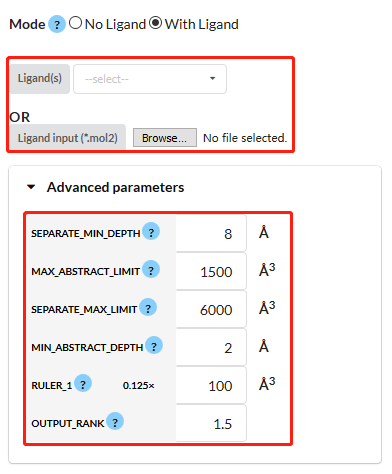
Figure 3. Visualization of "ligand mode" and "Advanced parameters" with red rectangles.
Step 4
Advanced parameters (shown in Figure 3). We have a set of default parameters to run CAVITY. And you are allowed to adjust some parameters of your interest. Explanations for the parameters:Step 5
Run CAVITY by clicking "Submit" button.
Table 1. Statistical analysis of Cavity computing time.
| Number of residues in protein | Predicted Running time(Unit: minute) |
|---|---|
| <400 | Usually less than 3 minutes |
| 400-500 | 2-5 |
| 500-600 | 4-7 |
| 600-800 | 6-9 |
| 800-1000 | 9-15 |
| >1000 | Usually longer than 15 minutes. The larger the protein, the longer the running time. |
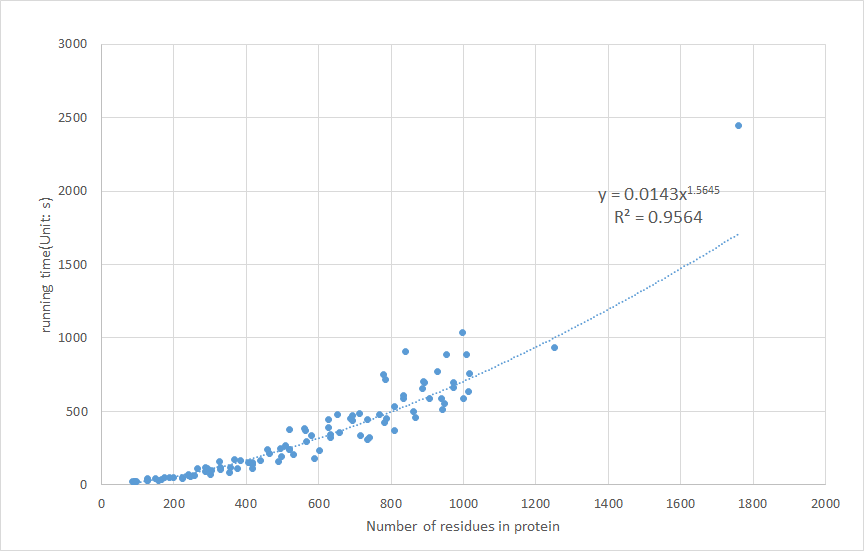
Figure 4. The scatter plot and fitted curve of Cavity processing time against the number of protein.
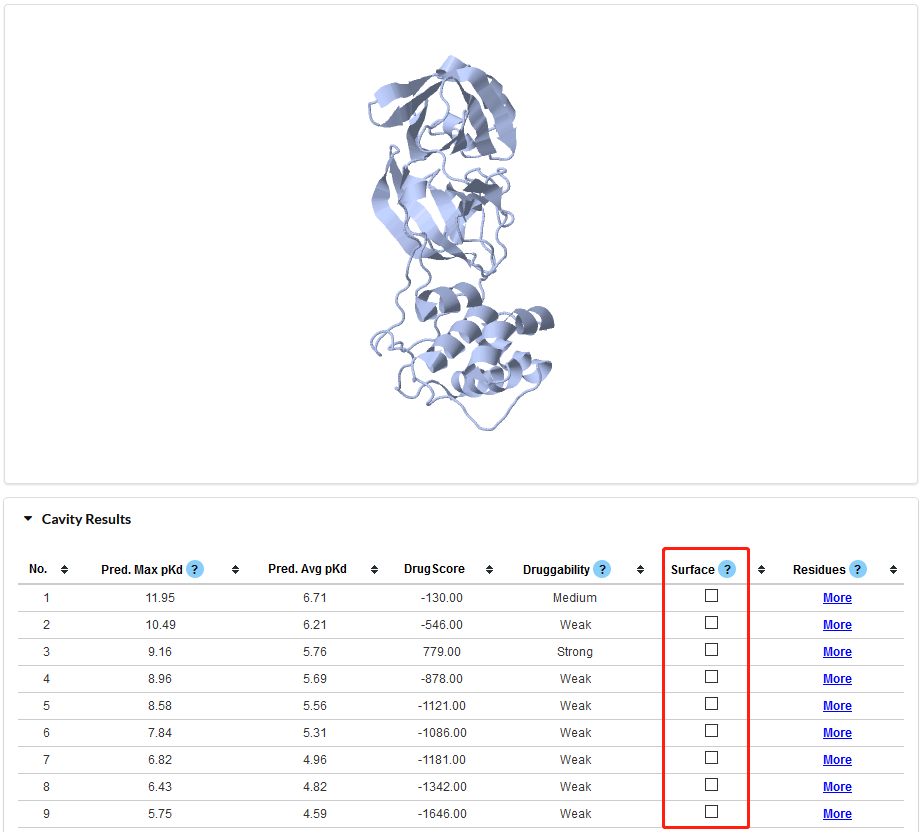
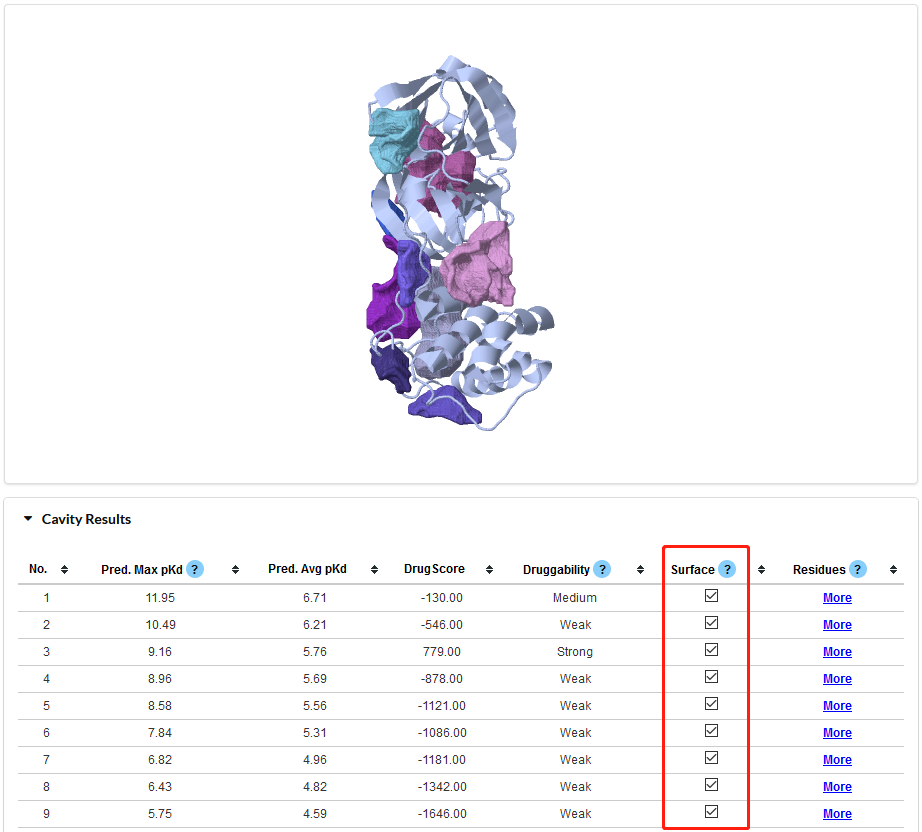
Figure 5. Visualization of Cavity Results. Once a checkbox in the Red rectangle is selected, the JSmol window will show the current cavity.
- name_surface.pdb: The output file storing the surface shape of the binding-site and the CavityScore. It is in PDB format, and user can use molecular modeling software to view this file and obtain an insight into the geometrical shape of the binding site. User can view this file by plain text editor, and check the predicted maximal pKd of the binding site. This value indicated the ligandability of the binding site. If it is less than 6.0(Kd is 1uM), suggests that this binding-site may be not a suitable drug design target.
- name_vacant.pdb: The output file storing the volume shape of the binding-site. It is in PDB format, and user can use molecular modeling software to view this file and obtain an insight into the geometrical shape of the binding site.
- name_cavity.pdb: The output file storing the atoms forming the binding-site. It is in PDB format, and user can use molecular modeling software to view this file and obtain an insight into the residues of the binding site. It is the visual version of "name_pocket.txt".
Back to top
CavPharmer
Step 1
To use CavPharmer, CAVITY module MUST be executed at first. When
Cavity finished successfully, a list of cavity results will be shown below the "Select a
cavity" label. Please select one result as input to generate pharmacophores; Once the radio button is selected, the JSmol window will show the residues of the selected cavity (shown in Figure 6).
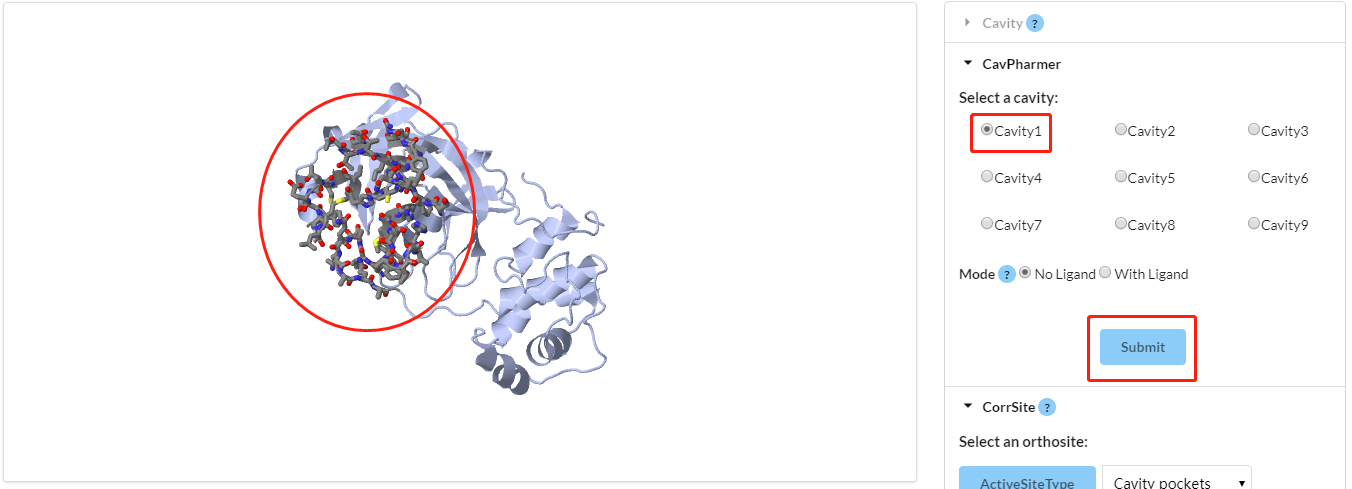
Figure 6. Visualization of CavPharmer operation interface. Red ellipse part shows the selected residues.
Step 2
Select mode.Step 3

Figure 7. Visualization of CavPharmer Results. Results can be recorded in the red rectangle sequentially.Different colors represent different pharmacophores. Blue and red spheres represent the H-bond donor center and h-bond acceptor, respectively; green spheres: hydrophobic center; olive spheres: positive electrostatic center; grey spheres: negative electrostatic center.
Back to top
CorrSite1.0
Step 1
Set the orthosteric sites:
1. Cavity pockets: results of CAVITY module. Select the one that you are interested.
2. Custom pockets: Upload one PDB file of orthosteric sites.
3. Custom residues: This server also support the custom .txt
file, like
the following formats: (residueID: ChainID)
46:A;47:A;49:A
or
46:A
47:A
49:A
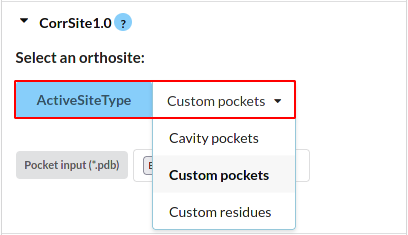
Figure 8. Visualization of CorrSite input format.
Step 2
Run program by clicking "Submit" button (It will takes only less than 30s).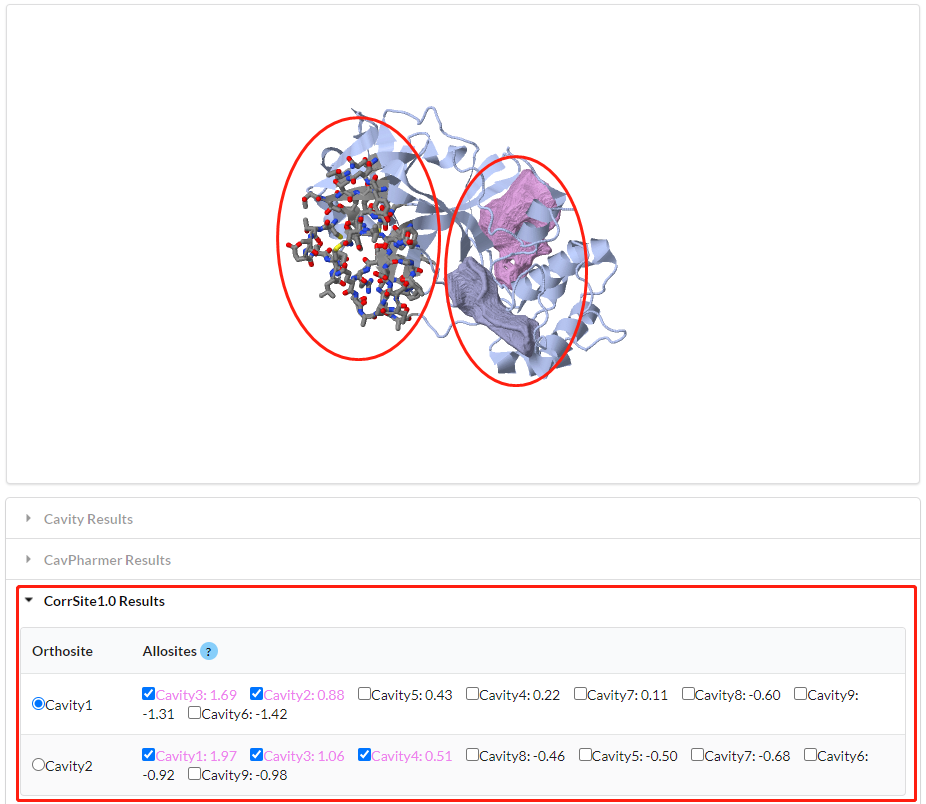
Figure 9. Visualization of CorrSite1.0 results. The stick region on the protein marked in red ellipse is the orthosteric site. The other region is corresponding to two allosteric sites. The score marked in a red rectangle in the "CorrSite1.0 Results" is a correlation score with the orthosite for the current cavity. When the score more than 0.5 (in pink color), this cavity may be a potential allosteric site.
Back to top
CorrSite2.0
CorrSite2.0 is a new method for predicting allosteric sites, which ranks the potential ligand binding sites based on calculated motion correlations using the slow and fast modes.
Step 1
Set the orthosteric sites:
1. Cavity pockets: results of CAVITY module. Select the orthosteric pocket that you are interested in, such as Cavity1.
2. Custom pockets: Upload one PDB file of orthosteric sites. For example, you can define the orthosteric pocket as all the residues within 4Å around the orthosteric ligand. We recommend you to use this option when the orthosteric pocket found by CAVITY is very large.
3. Custom residues: This server also support the custom .txt
file, like
the following formats: (residueID: ChainID)
46:A;47:A;49:A
or
46:A
47:A
49:A
In the ExcludeOrthoSite option, you can select the orthosteric pocket found by CAVITY to exclude the orthosteric pocket and predict potential allosteric sites in the remaining pockets. When the orthosteric pocket found by CAVITY is not very large, we recommend that you exclude the orthosteric pocket. When the orthosteric pocket found by CAVITY is very large, we recommend that you do not exclude the orthosteric pocket. If you are not sure whether to exclude the orthosteric pocket, you can directly select none of the pockets.
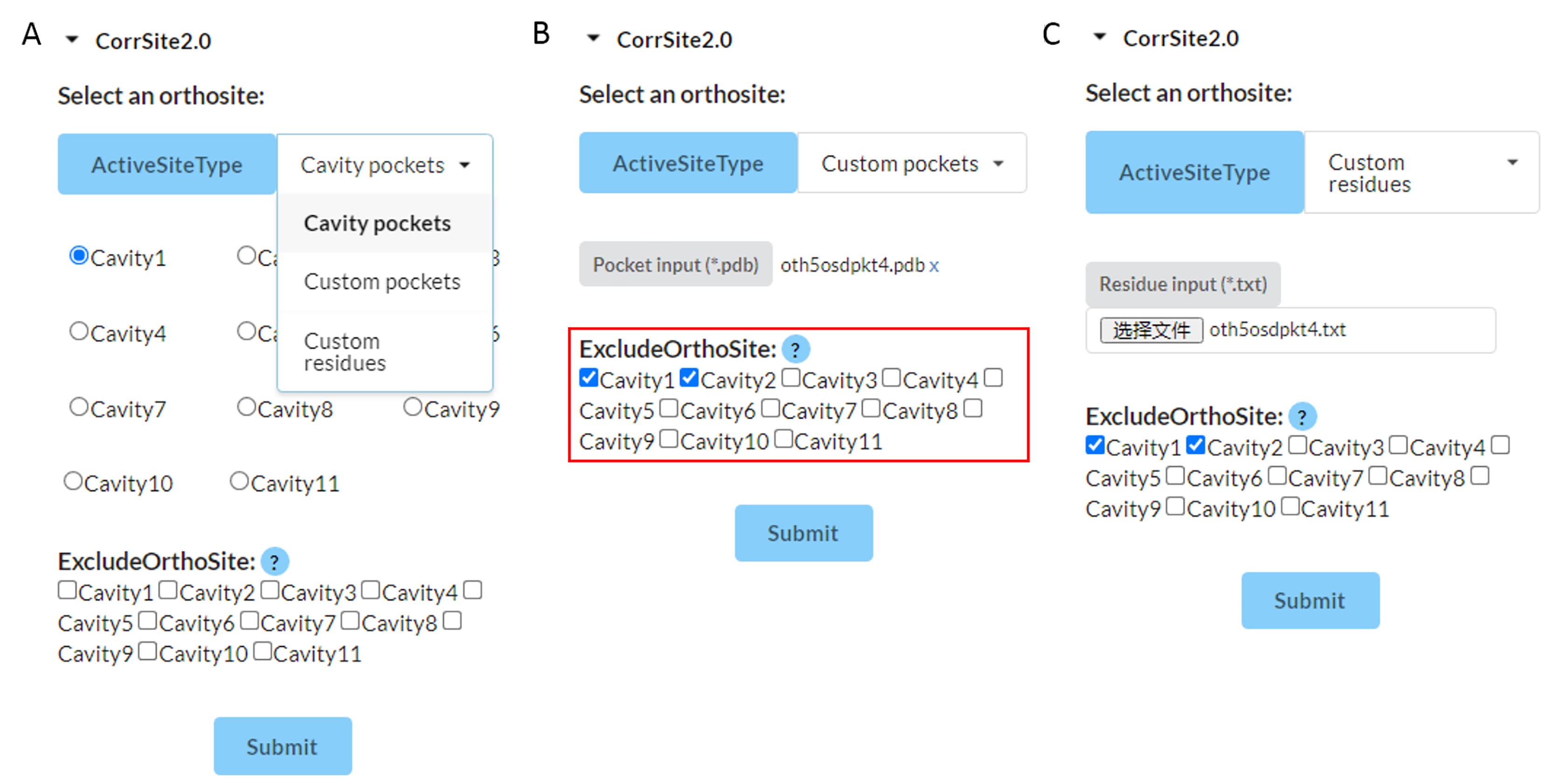
Figure 10. Visualization of CorrSite2.0 input format.
Step 2
Run program by clicking "Submit" button (It will takes only less than 30s).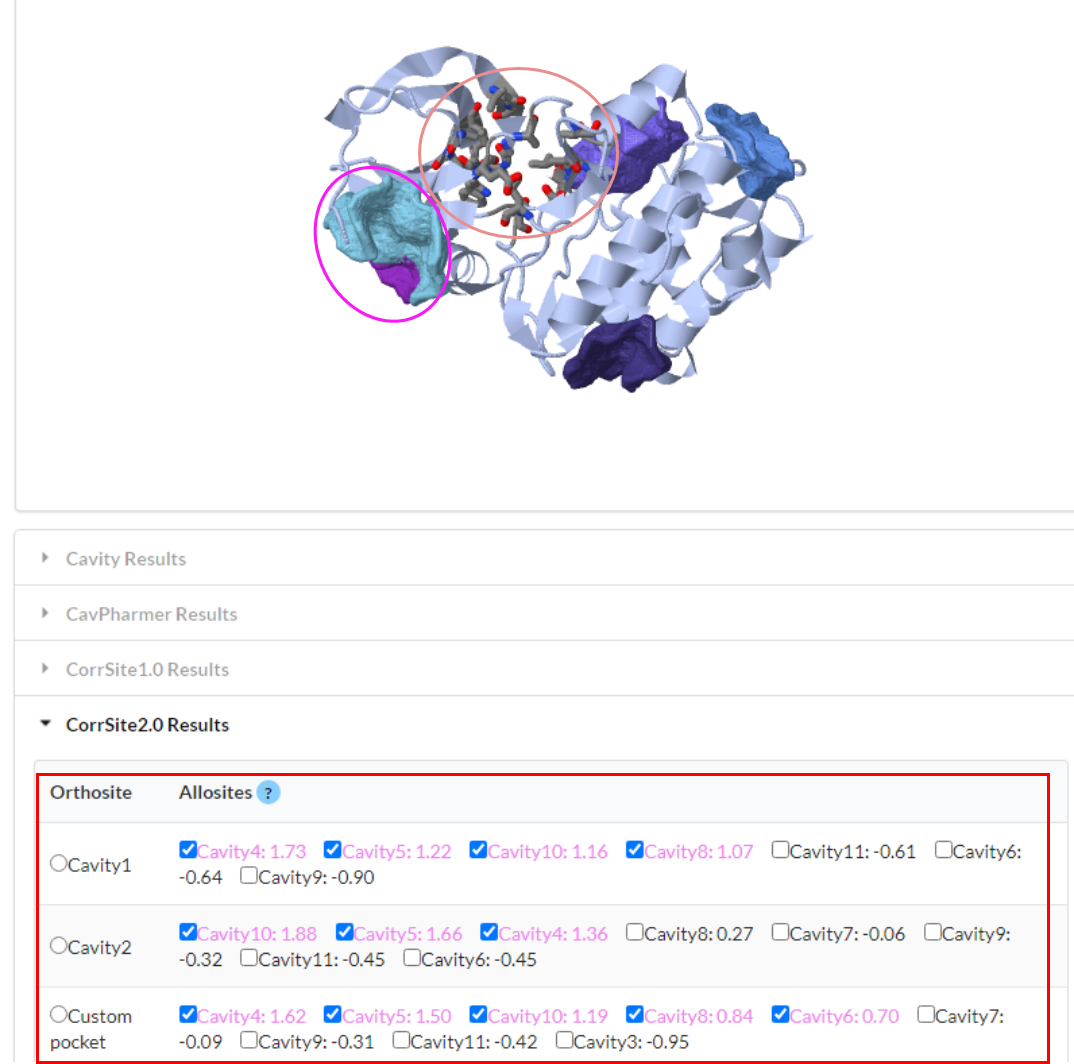
Figure 11. Visualization of CorrSite2.0 results. The stick region on the protein marked by pink ellipse is the orthosteric site. The other surface regions are corresponding to predicted potential allosteric sites, such as the region marked by magenta ellipse is the predicted allosteric pocket cavity4. The score marked in a red rectangle in the "CorrSite2.0 Results" are the correlation scores of the selected orthosteric site and other pockets. When the score more than 0.5 (in magenta color), this pocket may be a potential allosteric site.
Back to top
CovCys
Step 1
The input of this module are all the cavity results detected by the CAVITY program. Run
CAVITY first, then click "Run CovCys" button, the results will be shown the "CovCys Results"
part.
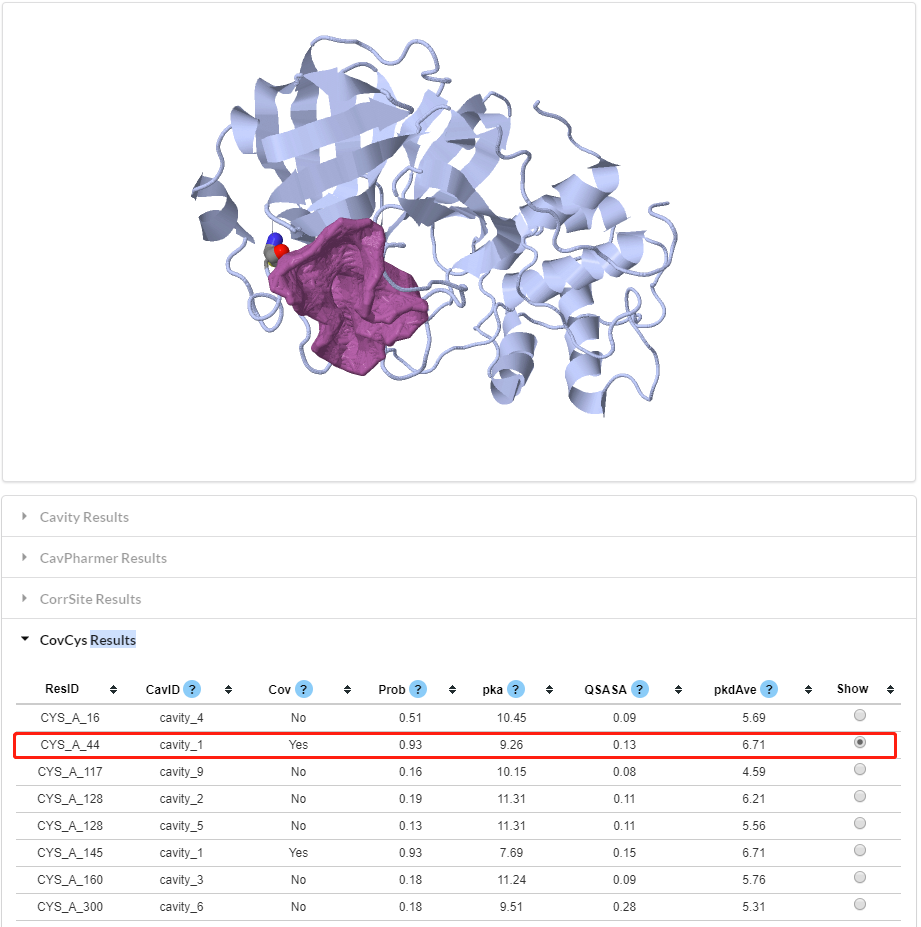
Figure 12. Visualization of CovCys results. Some key features are shown in the above table.
Download
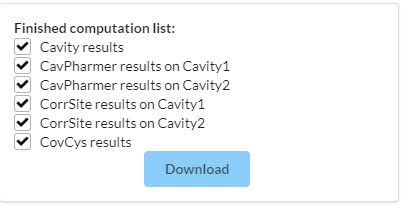
Figure 13. Visualization of Download list. The "Download" button is used to download the results of finished tasks.
Back to top
